如果您无法下载资料,请参考说明:
1、部分资料下载需要金币,请确保您的账户上有足够的金币
2、已购买过的文档,再次下载不重复扣费
3、资料包下载后请先用软件解压,在使用对应软件打开
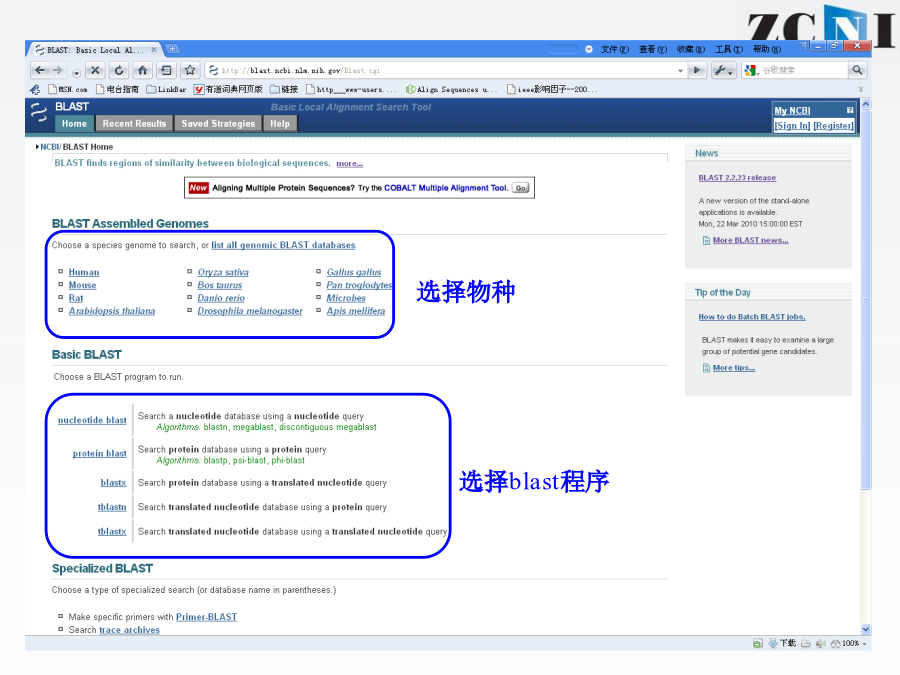
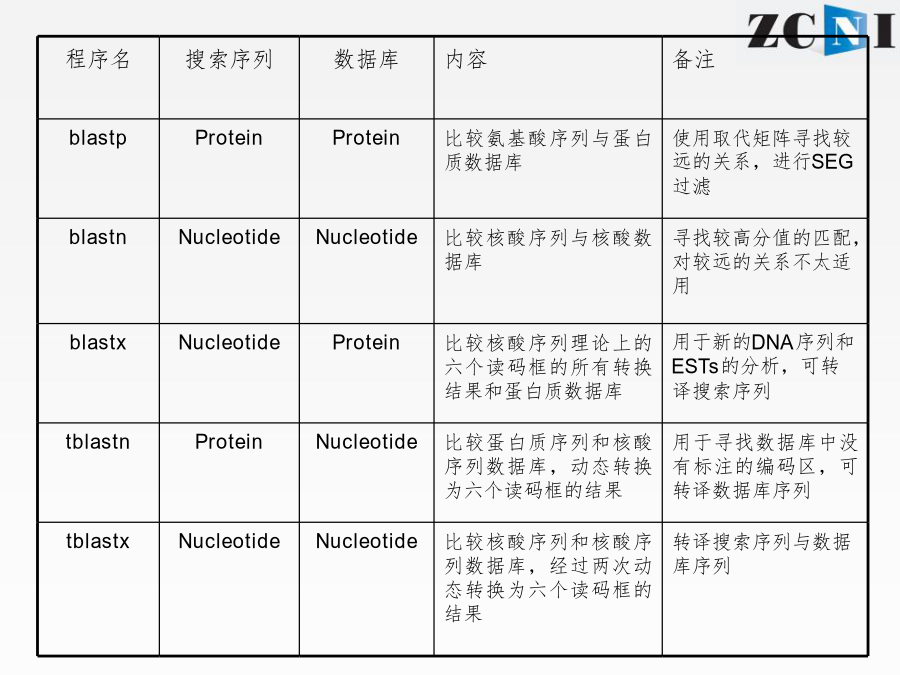
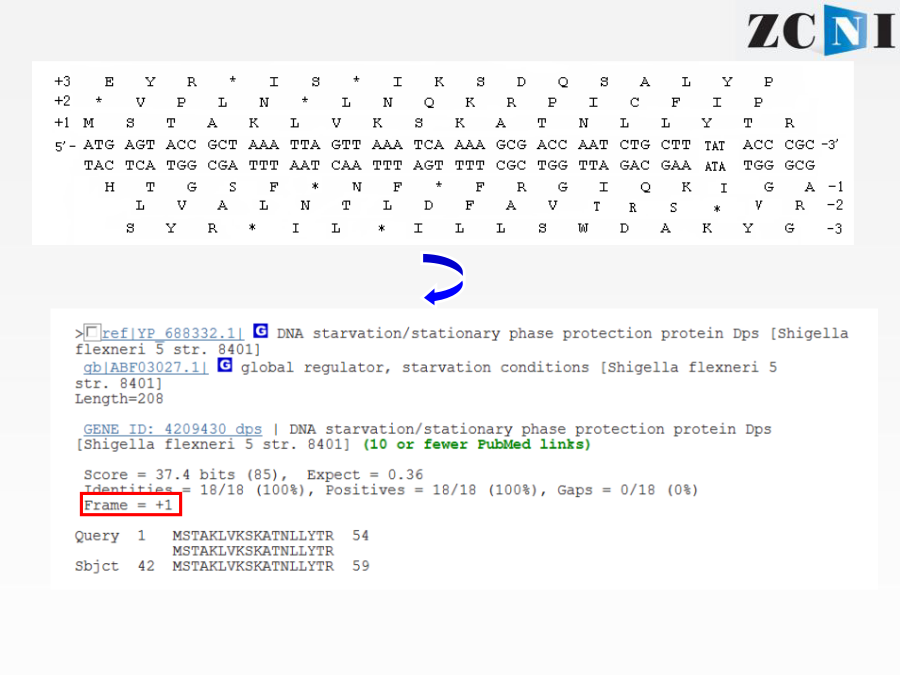
实习一基因组数据注释和功能分析实习一通过序列比对工具BLAST学习,了解蛋白编码基因的功能注释原理介绍多序列联配工具ClustalX分子进化分析软件MEGA5的基本知识,掌握系统发生树绘制的基本方法序列比对的进化基础BLASTQuerySequence程序名以Blastx为例:选择数据库blastp打分矩阵:PAM30PAM70BLOSUM80BLOSUM62BLOSUM45选择打分矩阵(scoringmatrix)比对的数据库信息E值(E-value)表示仅仅因为随机性造成获得这一比对结果的可能性。这一数值越接近零,随机发生这一事件的可能性越小,结果可靠性越高。blastn结果上机实习1:网上运行blastx和blastn本地运行BLAST登陆NCBI的FTP下载blast程序双击安装到C盘产生三个文件夹bindatadoc本地数据库的构建数据库的格式化程序运行上机实习2:本地运行blastx输入“cd\”,回车回到安装目录C盘输入“dir”,回车查看bin文件夹下内容输入“formatdb-idb-pT”,回车对db数据库进行格式化输入“dir”,回车察看bin文件夹下内容输入“blastall-pblastx-iin-ddb-oout-e2e-5-m9”-〉回车运行blastx程序产生的结果文件“out”用记事本查看结果文件不使用-m参数或者-m参数为0时显示序列两两比对结果多序列比对的目的ClustalW/X的运行目标序列点击StartJalview打开java程序窗口上机实习3:本地运行ClustalX在C:\zcni\shixi1\Clustalx2文件夹下,找到clustalx.exe双击打开ClustalX窗口点击File下拉菜单中Loadsequences选项,打开序列文件17-RNASE1.fasta.txt打开后的界面可在Alignment下拉菜单中的AlignmentParameters中设定各个参数点击进行多序列比对比对结果“*”、“:”、“.”和空格依次代表改位点的序列一致性由高到低MEGA5MEGA5可以识别fasta格式文件将17-RNASE1.fasta.txt重命名为17-RNASE1.fasta选择打开方式为MEGA5,打开17-RNASE1.fasta,自动跳出序列窗口用ClustalW做多序列联配ClustalW参数设置多序列联配后结果以.meg格式保存结果回到MEGA主窗口打开所保存的文件(.meg)点击按钮打开文件窗口显示保守位点显示变异位点回到MEGA主窗口构建进化树选择Bootstrap检验五种系统进化树构建方法进化树的可靠性分析软件上机练习4:MEGA5谢谢!